Меню
Меню





 Все темы
Все темы
Влияние синдрома Ретта на раннее развитие мозга зависит от типа вызвавшей его мутации
Причина развития синдрома Ретта — прогрессирующего расстройства центральной нервной системы — это мутации в гене белка MeCP2. Исследователи из США получили органоиды коры мозга из клеток двух пациентов с синдромом Ретта, имеющих миссенс-мутацию R306C или нонсенс-мутацию V247X, и проверили, как они повлияют на рост и работу органоидов. Органоиды моделируют раннее нейроразвитие пациентов. Мутация V247X сильнее влияла на функциональные реакции и связность, чем R306C. Выявленные на органоидах закономерности нашли отражение на ЭЭГ пациентов.
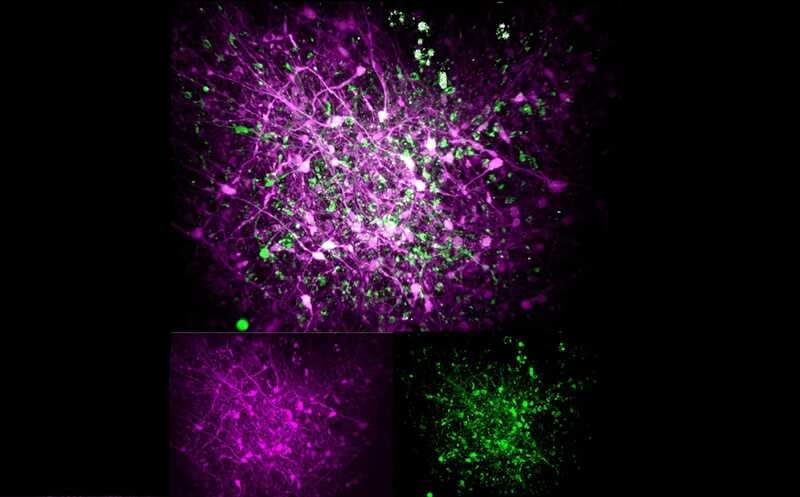
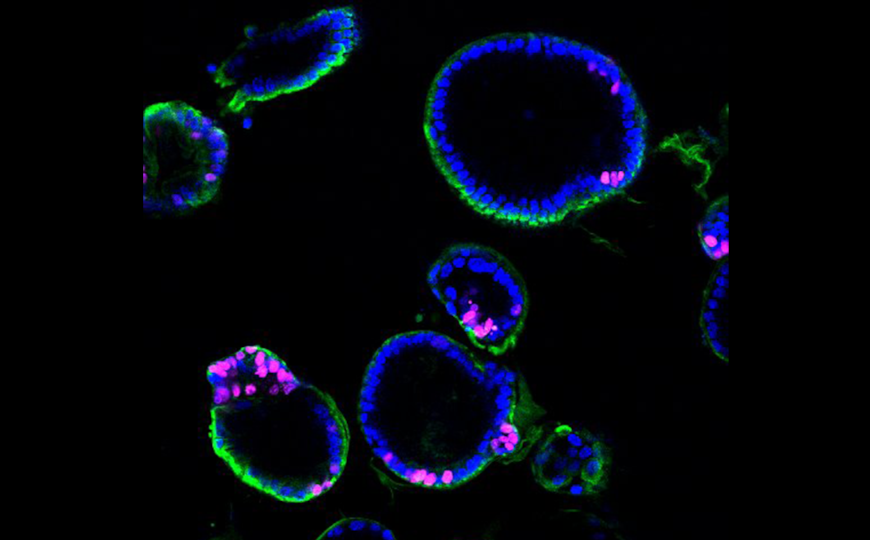
Credit: Tatsuya Osaki/MIT Picower Institute | Пресс-релиз
Синдром Ретта — прогрессирующее расстройство центральной нервной системы, вызванное мутациями в X-сцепленном гене, кодирующем метил-CpG-связывающий белок 2 (MeCP2); затрагивает в первую очередь девочек. Симптомы появляются в 6–18 месяцев, их тяжесть варьирует у пациентов и зависит от мутаций, вызвавших синдром Ретта. Были идентифицированы 863 уникальные мутации, но восемь из них составляют более 60% всех задокументированных случаев болезни.
MeCP2 напрямую связывает ДНК с помощью метил-CpG-связывающего домена (MBD) и привлекает корепрессоры или коактиваторы через C-концевой домен транскрипционного подавления (TRD). Тяжесть заболевания связана с типом мутации в дополнение к ее локализации в гене. Изучение взаимосвязи между типами мутаций MeCP2 и тяжестью заболевания важно для понимания молекулярных механизмов, лежащих в основе патологии синдрома Ретта, а также для разработки таргетированного лечения. Исследователи из США выяснили, как две разные мутации MeCP2 (916C>T / R306C и 705delG / V247X), расположенные близко друг к другу в TRD, влияют на структуру и функцию органоидов коры мозга, полученных из клеток пациентов. Эти органоиды отражают процессы, которые происходят на ранних этапах нейроразвития.
Органоиды получили из индуцированных стволовых клеток пациентов, несущих мутации MeCP2[R306C] и MeCP2[V247X], одновременно получили изогенные контроли без мутаций. Через три месяца культивирования авторы провели секвенирование РНК единичных клеток и выявили различные их типы, включая клетки-предшественники нейронов, клетки радиальной глии и промежуточные клетки-предшественники.
Диаметр органоидов V247X был значительно больше, чем контролей, чего не наблюдалось у органоидов R306C. Иммуногистохимическое криосекционирование выявило структурные различия в слоях нейронов. Органоиды V247X демонстрировали более тонкий слой CTIP2 и более толстый слой SATB2 по сравнению с изогенными контролями, тогда как органоиды R306C такой разницы не показали. При культивировании на плашках, покрытых матригелем, органоиды V247X продемонстрировали значительно меньшую толщину и длину аксонов, что указывает на более медленное созревание нейронов, в то время как органоиды R306C показали меньшую толщину аксонов по сравнению с контролем.
Авторы выявили значительное снижение средней активности в органоидах R306C по сравнению с изогенным контролем. В органоидах V247X средняя частота спайков статистически значимо снизилась, аналогично органоидам R306C. Кроме того, оказалось, что органоиды с мутациями R306C и V247X имеют пониженную синхронность.
В органоидах R306C на каждый нейрон приходилось больше связей, а в органоидах V247X обнаруживались отдельные нейроны с повышенным числом связей.
Исследователи сняли ЭЭГ у пяти пациентов с мутациями MeCP2[R255X], MeCP2[R270X] и MeCP2[R306C], а также у здоровых контролей того же возраста. В случаях с мутациями R255X и R270X наблюдалась примерно та же корреляция между различными участками мозга, что и у контролей, тогда как активность мозга у пациента с мутацией R306C, по-видимому, сильнее коррелировала в различных участках мозга по сравнению с контрольной группой. Были выявлены и другие различия, отражающие закономерности, показанные на органоидах.
РНК-секвенирование органоидов показало, что наличие мутации R306C повлияло на сигнальные пути, связанные с комплексами рецепторов, нейрональными шипиками, пептидил-тирозиновыми модификациями и биогенезом, включая пролиферацию и дифференцировку. Был выявлен дефицит комплексов клеточной адгезии, поддерживающих синаптические связи, особенно компонентов сигнального пути нейрексина-нейролигина и глутамата-рецептора глутамата, в органоидах с мутациями R306C по сравнению с контрольными.
В нейронах с мутацией V247X было меньше незрелых нейронов и астроцитов, чем в нейронах R306C или в контролях. Количество связей и сила нескольких путей нейрексина-нейролигина и ГАМК-ГАМК-рецепторного комплекса снижались (в частности, NRXN1-NLGN4X, NRXN1-NLGN3X и GABA-GABRA4).
Важную роль в дерегуляции числа связей играли астроциты. Так, взаимодействие астроцитов с возбуждающими нейронами и взаимодействие астроцитов с незрелыми возбуждающими нейронами в органоидах V247X было нарушено. Вероятно, дело в снижении экспрессии NRG3 и GLS в астроцитах и экспрессии ERBB4 как в ингибиторных, так и в возбуждающих нейронах.
В органоидах V247X наблюдалось снижение силы возбуждающе-тормозной связи, в то время как возбуждающе-возбуждающая связь усиливалась по сравнению с контролями. В органоидах R306C существенных различий в связности не наблюдалось.
Таким образом, как миссенс, так и нонсенс-мутации в гене, кодирующем MeCP2, могут повлиять на экспрессию генов, активность нейронов и работу их сетей. Влияние может быть как специфичным для мутации, так и общим для всех мутаций. Это важно для понимания мутационно-специфических механизмов развития синдрома Ретта и для разработки таргетных терапевтических средств.
Мишенью для лечения синдрома Ретта может быть микроглия
Источник:
Tatsuya Osaki, et al. Early differential impact of MeCP2 mutations on functional networks in Rett syndrome patient-derived human cortical organoids // Nature Communications (2026), published 14 April 2026, DOI: 10.1038/s41467-026-71458-0



 0
0